- Z Essaadi MD#essaadi dot z at gmail dot comService d’onco-radiothérapie, CHU Mohemmed VI, Marrakech, Maroc
- A Elomrani MDService d’onco-radiothérapie, CHU Mohemmed VI, Marrakech, Maroc
- M Khouchani MDService d'Onco-radiotherapie CHU Mohammed VI Marrakech Maroc
Le sarcome d’Ewing est une tumeur maligne touchant essentiellement le sujet jeune de moins de 20 ans. L’atteinte de la voute crânienne est rare, elle constitue 1% de toutes les localisations [1]. Nous rapportons une nouvelle observation afin d’illustrer la rareté et de mettre en évidence les particularités cliniques, thérapeutiques et évolutives de cette affection.
Il s’agit d’un adolescent âgé de 17 ans n’ayant pas d’antécédents pathologiques particuliers qui a consulté en aout 2009 pour une masse occipitale indolore augmentant progressivement du volume. Le scanner cérébral a objectivé un processus tumoral centré sur la voute crânienne temporale postérieure gauche, compliqué d’un envahissement des parties molles pariétales et du parenchyme cérébral en regard (figure 1). Une exérèse partielle de la masse a été réalisée et l’étude histologique a objectivé une prolifération maligne à cellules rondes (PNET, rhabdomyosarcome, autres), un complément immuno histochimique a confirmé le diagnostic de sarcome d’Ewing (CD56+, CD3-, CD20-, CD45-, myogenin-, myélopéroxidase-, protéine S100-). Un scanner thoracique et une échographie abdominale ont été faits dans le cadre de bilan d’extension, qui sont normaux. Le patient a bénéficié de 6 cures de chimiothérapie type VAC (vincristine, adriblastine, cyclophosphamise) /IE (Iphosphamide, étoposide) puis radiothérapie à la dose de 50gy en fractionnement classique (figure 2) puis 6 cures de VAC (vincristine, dactinomycine, cyclophophamide)/IE, l’évolution a été marquée par une rémission complète jusqu’à 2012, le malade a consulté pour une masse dure au niveau de la cicatrice opératoire d’apparition récente prurigineuse. Un scanner cérébral a été réalisé et qui a objectivé un aspect en faveur d’une récidive locale. Le patient a été opéré, le geste avait consisté à une exérèse de la masse tumorale. A l’étude histologique c’était un aspect en faveur de sarcome d’Ewing déjà diagnostiqué initialement. Le malade a bénéficié de 4 cures de chimiothérapie complémentaire bien tolérée à base d’adriablastine et iphosphamide. Actuellement il est en rémission avec un recul d’une année.

Le sarcome d’Ewing est la tumeur maligne la plus fréquente de l’os après l’ostéosarcome chez le sujet jeune [1]. C’est l’apanage de l’enfant et l’adulte jeune de moins de 20 ans avec un pic d’incidence entre 5 et 13 ans. Une prédominance masculine a été notée avec un sex- ratio de 1, 6 [2] il atteint par ordre de fréquence les os longs (47%), le pelvis (19%) et les cotes dans 12% des cas. Il concerne le plus souvent le membre inférieur que le membre supérieur [3] [4]. l’atteinte primitive de la voute crânienne reste exceptionnelle et constitue 1% de toutes les localisations. Le sarcome d’Ewing a été décrit la 1ere fois par James d’Ewing, c’est une tumeur à cellules rondes, qui appartient à la famille des tumeurs neuroectodermiques qui ont en commun une translocation cytogénétique entre le chromosome 22 et 11 dans la majorité des cas [5] [6]. D’habitude, la voute crânienne est le siège de métastases, une atteinte primitive est rare. La localisation frontale et pariétale est la plus fréquente [7]. Néanmoins la localisation temporale et occipitale a été également rapportée [8] [9].
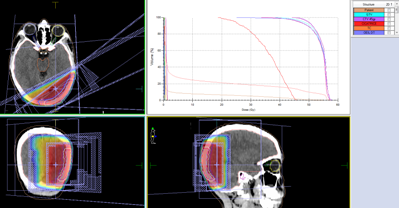
Cliniquement, les symptômes sont très variés et hétérogènes mais non spécifiques. Habituellement la masse et la tuméfaction de la voute crânienne d’apparition récente avec l’augmentation rapide de volume est le signe révélateur, Des céphalées voire un syndrome d’hypertension intracrânien en cas d’extension de la tumeur en intra crânien et enfin une altération d’état général peut être observée. Sur le plan radiologique, une radiographie standard du crane permet de mettre en évidence des lésions osseuses sans pouvoir faire la nuance entre une lésion maligne ou bénigne ainsi c’est très difficile de distinguer un sarcome d’Ewing d’un lymphome de burkitt, d’un kyste anévrismal ou d’une dysplasie fibreuse. Le scanner cérébral avec une fenêtre osseuse reste l’examen du choix, et qui permet une meilleurs analyse des lésions osseuses et l’extension intra et extra crânienne. Il met en évidence une masse épidurale iso ou hyperdense se rehaussant de façon hétérogène après injection du produit de contraste avec ostéolyse et envahissement du cuir chevelu [1] [2]. L’imagerie par résonance magnétique permet d’objectiver avec précision la taille tumorale, les rapports anatomiques avec les structures cérébrales et vasculaires ainsi que l’extension de l’œdème péri lésionnel. Les sarcomes d’Ewing sont des tumeurs de bon pronostic. la prise en charge des sarcomes d’Ewing repose sur la chimiothérapie et la chirurgie voire une radiothérapie en cas d’exérèse partielle ou tumeur inopérable. La chimiothérapie a pour objectif de prévenir l’apparition des métastases dans les formes localisées ou de les traiter si le malade est métastatique d’emblée. les médicaments ayant montré une efficacité en mono chimiothérapie sont la vincristine, le cyclophosphamide, l’actinomycine D et l’adriamycine. L’association de ces médicaments se fait selon différents régimes. L’adjonction d’ifosfamide et d’étoposide est utile dans les grosses tumeurs [10]. Les taux de survie restent toujours faibles dans les formes métastatiques ne dépassant pas 20% et ce malgré une bonne réponse à la chimiothérapie initiale, et afin d’améliorer ces résultats, la chimiothérapie intensive suivie de greffe de cellules souches hématopoïétiques est préconisée [10]. La chirurgie est le traitement du choix des formes localisées après une chimiothérapie afin d’assurer le contrôle local. Concernant la radiothérapie, la dose recommandée est de 55 à 60 gy. Une adaptation de la dose est préconisée selon le siège, le volume et la proximité d’organes sensibles. La radiothérapie est administrée après un acte chirurgical si les marges d’exérèses sont tumorales et le chirurgien a jugé l’impossibilité de prendre une large marge autour de la tumeur. Sinon on a recours à la radiothérapie plutôt qu’à la chirurgie si la tumeur est jugée inopérable [10].
L’évolution de cette affection peut marquer par l’apparition des métastases osseuses, pulmonaires et cérébrales. La survie à 5 ans est estimée de 50 à 80% [2].
le sarcome d’Ewing est une tumeur maligne du sujet jeune. La localisation crânienne est rare. L’absence de récidive et des métastases et une prise en charge précoce et bien codifiée rendent le pronostic de ces tumeurs bon.
- Bricha M, Jroundi L, Boujida N, El Hassani M, Chakir N, Jiddane M. [Primary Ewing sarcoma of the skull vault]. J Radiol. 2007;88:1899-901 pubmed
- Gadani S, Mody RP, Solanki RN, Mahajan A. Primary Ewing sarcoma on skull vault in a child. Ind J Radiol Imag 2003;13:303-5.
- Yih L, Windy YC, Taipei. Ewing’s sarcoma of the hand. J Hand Surg 1998;23A:748–52.
- Dreyfuss U, Auslander L, Bialik V, Fishman J. Ewing's sarcoma of the hand following recurrent trauma; a case report. Hand. 1980;12:300-3 pubmed
- Desai K, Nadkarni T, Goel A, Muzumdar D, Naresh K, Nair C. Primary Ewing's sarcoma of the cranium. Neurosurgery. 2000;46:62-8; discussion 68-9 pubmed
- Roosen N, Lins E. Primary Ewing's sarcoma of the calvarial skull. Neurochirurgia (Stuttg). 1991;34:184-7 pubmed
- Özdemir N, Tektas S. The calvarial lesions. Journal of Neurological sciences Turkish 2004;21:57-75.
- Yamashita Y, Kumabe T, Kobayashi T, Abiko H, Seki H, Yoshimoto T. [Ewing's sarcoma at the occipital bone presenting as acute epidural hematoma: a case report]. No Shinkei Geka. 1997;25:567-71 pubmed
- Zenke K, Hatakeyama T, Hashimoto H, Sakaki S, Manabe K. Primary Ewing's sarcoma of the occipital bone--case report. Neurol Med Chir (Tokyo). 1994;34:246-50 pubmed
- In: le cancer chez L’enfant; aspects pratiques. Harif M. Imprimerie l’union –juillet 2012.