- Caroline Ritchie Ph. D.caroline dot ritchie103 at gmail dot com
- Galet Colette Ph. D.cogalet at gmail dot comUniversité de Californie à Los Angeles, États-Unis
Les protéines purifiées sont nécessaires pour de nombreuses applications expérimentales, y compris des études structurelles et des dosages biochimiques in vitro. Les protéines peuvent être obtenues à partir de tissus ou, plus souvent, par leur surexpression dans un organisme modèle, comme des bactéries, des levures ou des cellules de mammifères en culture. La purification des protéines consiste à isoler des protéines, sur la base de différences dans leurs propriétés physiques. L'objectif d'un système de purification des protéines est de conserver la plus grande quantité de protéines fonctionnelles avec moins de contaminants possible. Le système de purification d'une protéine doit être optimisé pour achever ce processus dans le plus petit nombre d'étapes.

L'article passe en revue quatre types de chromatographie sur colonne qui sont couramment utilisés dans la purification des protéines, et discute les avantages, les inconvénients et les problèmes potentiels de chacun. Cet article rapporte également une revue de Labome sur 98 publications. La revue indique que la chromatographie sur colonne d'affinité, surtout basée sur les tags HIS, GST, et FLAG tags, et la chromatographie d'exclusion stérique sont les principales méthodes citées dans les publications. GE Healthcare est le principal fournisseur de réactifs et d'instruments utilisés dans la purification des protéines.
Le facteur le plus important dans le développement d'un système de purification des protéines est l'application en aval de la protéine purifiée. La quantité et la pureté de la protéine doit être suffisante pour l'analyse expérimentale. De plus, des informations sur le comportement de la protéine doivent être prises en considération, comme une protéine dons sa conformation native et fonctionnelle est nécessaire pour les études en aval. Pendant la purification et le stockage ultérieur, de nombreux processus peuvent se produire qui affectent la qualité des protéines: la dérnaturation des protéines, l'agrégation, la dégradation et la perte de fonction. Une planification minutieuse pour purifier une protéine aussi rapidement que possible et dans les conditions les plus stables permettra de maximiser les chances d'un système de purification efficace.

Les conditions de la solution d'une protéine à chaque étape du schéma de purification sont essentielles dans le maintien de la stabilité et de la fonction des protéines. Les protéines doivent être conservées dans un environnement bien tamponné pour éviter les changements brusques de pH qui pourraient affecter de manière irréversible leur repliement, leur solubilité, et leur fonction.
Un tampon est une solution contenant une paire d'acide / base conjugués. La gamme de pH d'un tampon repose sur so pKa, défini comme le pH à laquelle 50% des molécules sont sous leur forme acide, et 50% sont sous leur forme basique (figure 1). Une règle générale en ce qui concerne les tampons est que le pH de la solution tampon devrait être dans 1,0 unité de pH du pKa de manière à fournir une capacité tampon appropriée. Ceci garantit qu'il existe une quantité suffisante de la molécule sous ses deux formes acide et basique pour neutraliser la solution dans le cas d’un afflux de H+ ou OH- Ainsi, les tampons empêchent les changements de pH qui pourraient affecter négativement la stabilité des protéines.

Un bon tampon doit présenter les caractéristiques suivantes [1] :
- Solubilité dans l'eau
- Stabilité chimique
- Grande capacité tampon au pH désiré
- Compatibilité avec les applications analytiques et expérimentales
- Compatibilité avec les autres composants de la solution
De nombreux éléments peuvent servir de tampons biologiques. Comme les tampons peuvent être utilisés à un pH physiologique, les composants de tampon les plus couramment utilisés ont un pKa, proche de la neutralité. Quatre des tampons biologiques les plus courants sont énumérés dans la table 1, ainsi que la gamme de pH à laquelle ils peuvent être utilisés, et les avantages et les inconvénients qui pourraient influer sur leur utilisation dans la purification des protéines. En règle générale, ces tampons sont utilisés à des concentrations supérieures à 25mm pour assurer la capacité de tampon adéquate.
| Tampon | Gamme de pH | Avantages et inconvénients |
|---|---|---|
| Phosphate | 5.8-8.0 |
|
| MOPS | 6.5-7.9 |
|
| HEPES | 6.8-8.2 |
|
| Tris | 7.5-9.0 |
En plus d'un système tampon approprié, les solutions utilisées au cours de purification de protéines, de la lyse de stockage, contiennent souvent de nombreux autres composants qui jouent un rôle dans la facilitation de la pureté, de la stabilité et de la fonction de la protéine.
Les inhibiteurs de protéase sont souvent ajoutés au tampon de lyse et dans les premières étapes du procédé de purification visant à prévenir la dégradation de la protéine cible par des protéases endogènes. Ceux-ci ne sont généralement pas nécessaires dans les stades ultérieurs de la purification, comme la plupart ou la totalité des protéases contaminantes ont été séparées de la protéine d'intérêt. Les réactifs chélateurs de métaux tels que l'EDTA ou l'EGTA, sont souvent ajoutés au tampon de stockage. Ces chélateurs de métaux se lient Mg2+ et, ainsi, empêchent le clivage de la protéine purifiée par des métalloprotéases contaminantes. D'autres additifs sont souvent utilisés pour protéger les protéines contre les dommages et d'améliorer leur solubilité.
| Type | Fonction | Réactifs généralement utilisés |
|---|---|---|
| Agents réducteurs | Protègent contre les dommages liés à l’oxydation |
|
| Inhibiteurs de protéases | Inhibent la dégradation des protéines par les protéases endogènes |
|
| Chélateurs de métaux | Inactive les métalloprotéases | |
| Osmolytes | Stabilise la structure protéique et augmente la solubilité |
|
| stabilisateurs ioniques | augmente la solubilité | Sels (ex., NaCl, KCl, (NH4)2SO4 |
Les additifs ne doivent être utilisés que si nécessaire. Les étapes d’essais et d’erreurs sont souvent nécessaires pour déterminer les additifs spécifiques qui sont bénéfiques à la purification d’une protéine particulière.
D'autres facteurs contribuent également à la stabilité de la protéine lors de la purification. Le moins de manipulation possible lors de la purification d’une protéine est toujours la meilleure approche. La conception d'une méthode de purification qui utilise le nombre minimum d'étapes dans un laps de temps court assure le plus haut rendement de protéines fonctionnelles. De plus, il est souvent préférable de garder la protéine à froid tout au long de la purification. Typiquement, la purification est effectuée à 4°C, car cette température inférieure ralentit la vitesse de protéolyse (en cas de contamination de protéases) et favorise l'intégrité structurale des protéines.
Le principe de la chromatographie sur colonne est de séparer un pool de protéines en de nombreux petits pools, dont certains sont enrichis avec la protéine d'intérêt. Bien que l'équipement coûteux et spécialisé soit disponible pour chromatographie sur colonne, seul l'équipement de base est nécessaire.

L'équipement de base pour la chromatographie sur colonne:
- Phase stationnaire - une matrice inerte, souvent avec un groupe fonctionnel attaché pour faciliter l'interaction de la protéine, qui sert à séparer les protéines. Le choix de la phase stationnaire et le groupe fonctionnel dépend à la fois du type de chromatographie qui est en cours d'exécution et le procédé par lequel elle sera réalisée.
- Colonne -un réservoir cylindrique en verre disponible en différentes longueurs et diamètres. Les colonnes peuvent être achetées pré-remplies avec une phase stationnaire et prêtes à être attachées aux systèmes automatisés de chromatographie (discuté dans la partie 2 de cette section) ou peuvent être achetées vide pour le remplissage manuel. Différents types de colonnes sont nécessaires pour la chromatographie automatisée par rapport à la chromatographie de flux par gravité.
- Solvants - des tampons contenant des additifs utilisés pour l'équilibrage, le lavage, et l’élution de protéines de la phase stationnaire. Différents types de chromatographie nécessitent des conditions différentes de solvant.
- Tubes de prélèvement – Tubes utilisés pour recueillir les échantillons d'élution. Des tubes spéciaux sont nécessaires pour les collecteurs de fractions automatisés, mais n'importe quel tube est approprié pour la collecte de fractions manuelle.
- Test pour mesurer la pureté - une approche pour déterminer la quantité relative d'une protéine spécifique par rapport aux protéines totales dans un échantillon. Les fractions contenant la protéine d'intérêt doivent être déterminées après chaque étape avant de passer à la prochaine étape dans la méthode de purification. Certaines méthodes courantes d'analyse de pureté seront examinées dans une section ultérieure.
La chromatographie sur colonne peut être réalisée avec des systèmes automatisés (figure 2), qui utilisent une pompe pour forcer le solvant sur une colonne remplie de matrice à un débit consigne constant, ou peut être exécutée par gravité. Les deux systèmes, automatique et par gravité peuvent être couplés à des systèmes de collecte de fractions automatiques. Il y a des avantages et des inconvénients pour chaque système. Le système de GE Healthcare, FPLC AKTA, est un choix très commun [5] [6] [7] [8].
| Type de Système | Avantages | désavantages |
|---|---|---|
| automatisé |
|
|
| flux de gravité |
|
|
Les quatre principaux types de chromatographie sur colonne comprennent la chromatographie d'affinité, la chromatographie échangeuse d'ions (IEX), la chromatographie d'interaction hydrophobe (HIC), et la chromatographie d'exclusion de taille (SEC). La plupart des systèmes de purification nécessitent l'utilisation de plusieurs de ces types de procédures chromatographiques pour obtenir la pureté nécessaire pour les applications en aval. Le choix de la (les) méthode(s) chromatographique(s) la (les) plus appropriée(s) et l'ordre de ces méthodes est essentielle dans l'optimisation d'une méthode de purification des protéines.
| Type de Chromatographie | Sépare les protéines par | lie avec | Elue avec |
|---|---|---|---|
| Affinité | une interaction spécifique | pas de ligand de compétition | ligand de compétition (spécifique); conditions qui rompent les interactions protéine/protéine (non-spécifique) |
| Echange d’ions | Charge de surface nette | force ionique faible | Force ionique élevée, augmentant (échange de cations) ou diminuant (échanges d’anions) pH |
| Interactions Hydrophobe | Hydrophobicité | force ionique élevée | force ionique faible |
| Exclusion de taille | Radius Hydrodynamique |
En analysant la séquence d'une protéine, des caractéristiques uniques peuvent être identifiées qui peuvent aider à sa purification. La taille et la charge d'une protéine (à un pH spécifique) peut être déterminée, ainsi que l'identification de grandes régions de résidus hydrophobes.
La chromatographie d'affinité repose sur la fixation spécifique et réversible d'une protéine à un ligand lié à la matrice. Le ligand peut soit se lier directement à la protéine d'intérêt ou à un tag qui est attaché de manière covalente à la protéine. La chromatographie d'affinité est souvent la procédure de purification la plus robuste et est généralement utilisée dans les premiers stades de la méthode de purification. En fonction de l’application en aval, la purification par affinité peut être la seule étape de chromatographie nécessaire pour atteindre une pureté adéquate.

La phase stationnaire pour la chromatographie d'affinité est réalisée sur une matrice inerte fixée de manière covalente à un ligand qui se lie spécifiquement à une protéine ou un groupe de protéines. La matrice inerte est généralement composée d'agarose réticulé ou de polyacrylamide. Les protéines peuvent être purifiées par chromatographie d'affinité d'une manière sélective ou non sélective. Dans la chromatographie d'affinité sélective, un ligand spécifique d'une protéine ou d'un tag lié de manière covalente est utilisé. Dans la chromatographie d'affinité non sélective comme la protéine A, G, L pour les immunoglobulines, ou de l'héparine pour des protéines liant l'ADN, ou la lectine pour les glycoprotéines, le ligand se lie à un groupe de protéines avec des capacités de liaison similaires.
Dans les deux types de chromatographie d'affinité, les protéines sont chargées sur la colonne dans des conditions qui influencent la liaison entre la protéine (ou tag) et son ligand. La protéine liée est lavée dans des conditions qui ne perturbent pas l'interaction spécifique, mais qui peut perturber les interactions non spécifiques entre protéines contaminantes et la phase stationnaire. La protéine liée est ensuite éluée avec un tampon contenant une molécule concurrente ou des conditions qui perturbent les interactions protéine / protéine. Les molécules concurrentes se lient au ligand, déplaçant la protéine d'intérêt. Cette molécule concurrente est généralement éliminée de la protéine d'intérêt, soit par un autre procédé chromatographique ou une dialyse. Les méthodes d’élution des protéines à partir de la phase stationnaire en perturbant les interactions protéine / protéine incluent ajuster le pH ou la force ionique du tampon. Ces méthodes peuvent affecter la stabilité de la protéine, et il est suggéré de neutraliser ou de diluer la protéine éluée immédiatement pour minimiser les dommages. Pour certaines formes de chromatographie d'affinité, des conditions d'élution alternatives ont été décrites afin de maximiser le rendement de protéines fonctionnelle [11] [12].
| Protéine à purifier | Ligand | Eluée avec | |
|---|---|---|---|
| Anticorps (antigène-spécifique) | peptide antigénique | peptide libre | |
| Protéine taguée avec le tag Poly histidine | Ni2+ ou Co2+ | Imidazole ou histidine libre | |
| Protéine taguée avec l’épitope FLAG | Anticorps anti- FLAG spécifique | peptide FLAG ou pH faible | |
| Protéine taguée avec le tag GST | glutathion réduit | glutathion libre | |
| Protéine taguée avec l’épitope Myc | Anticorps anti-Myc spécifique | faible pH | |
| Anticorps (classe-spécifique) | Protéine A, G, ou L | pH extrêmes | |
| Protéine de liaison a l’ADN | Héparine | Force ionique élevée |
Dans la conception d'un plasmide pour l'expression des protéines, des tags d'affinité peuvent être insérés sur l'extrémité N-ou C-terminale (ou dans certains cas rares, dans des boucles flexibles de protéines ayant des structures connues) pour aider à la purification. Voir l’article de Labome sur les tags protéiques et peptidiques pour une liste de tags communs et une discussion de chacun.
Les anticorps sont purifiés souvent sur la base de l'interaction très spécifique qui se produit entre un anticorps et son antigène (la séquence reconnue par l'anticorps). Un peptide contenant l'antigène peut être couplé à une matrice pour une liaison spécifique de l'anticorps. L'abaissement du pH du tampon d'élution perturbe l'interaction anticorps / peptide pour libérer l'anticorps lié. Cette méthode est souvent utilisée dans le commerce pour l'isolement d'anticorps à partir du sérum brut.

Les protéines peuvent également être purifiées par affinité d'une manière non-sélective. Dans la purification non-sélective, le ligand fixé à la phase stationnaire se lie à un groupe de protéines avec des partenaires de liaison similaires. Un exemple de purification par affinité non sélective est la purification des protéines liant l'ADN. L’héparine imite l'ADN à la fois dans sa structure et sa charge, et peut être utilisée en tant que ligand pour la purification par affinité des protéines liant l'ADN. Alors que toutes les protéines liant l'ADN peuvent théoriquement se lier à cette phase stationnaire, la plupart des autres protéines ne le font pas et sont éliminées, conduisant à un enrichissement suffisant de la protéine d'intérêt. Un autre exemple est l'enrichissement des anticorps par la liaison de leur région constante (Fc) au ligand, la protéine A, G ou L. Les colonnes d'affinité protéine A de GE Healthcare sont un choix commun [13] [14] [15], comme l’est la protéine G [16].
L'utilisation d'un même mode de liaison (un anticorps lié à une protéine ligand A, G, ou L), la région de liaison à l'antigène (Fab) de l'anticorps est encore disponible pour la liaison à son antigène spécifique. Ainsi, certaines protéines peuvent être purifiées sur la base de l'interaction spécifique entre le ligand / anticorps couplé et l'antigène de l'anticorps.
Problèmes courants et solutions sont listés dans le Tableau 6.
| Problème | Cause | Solution |
|---|---|---|
| La protéinée ne se lie pas | Le tag n’a pas été traduit ou n’est pas accessible | vérifier la séquence du plasmide ou déplacer le tag sur une autre région |
| Les conditions de liaison ne sont pas appropriées | Ajuster les conditions des tampons | |
| Pas assez de temps pour la liaison | diminuer le débit ou arrêter la colonne pour permettre l’incubation | |
| La protéine n’est pas éluée | L’affinité entre le ligand et le tag est très élevée | Augmenter la concentration du compétiteur (spécifique) ou la stringence des conditions (non-spécifique) |
| les protéines se sont agrégées sur la colonne | Ajuster les conditions tampons pour une meilleure stabilité de la protéine | |
| résolution faible | le débit est soit trop rapide ou trop lent | Ajuster le débit |
| La colonne n’a pas été suffisamment lavée | Laver avec un tampon plus stringent, nettoyer la matrice selon les directions du fournisseur | |
| Les protéines se sont agrégées sur la colonne | Ajuster les conditions du tampon pour une meilleure stabilité des protéines | |
| Les conditions d’élution ne sont pas assez stringente | Augmenter les concentrations du compétiteur (spécifique) ou ajuster les conditions (non-spécifique) | |
| La protéine a perdu son activité pendant la procédure | La protéine est dépliée ou agrégée | Ajuster les conditions du tampon pour une meilleure stabilité de la protéine |
| Un cofacteur nécessaire pour l’activité a été éliminé pendant la purification | Ajouter le cofacteur |
La chromatographie par échange d'ions (IEX) sépare les protéines en fonction de leur charge de surface nette, grâce à des interactions électrostatiques qui se produisent entre les protéines et une phase stationnaire chargée. Deux types d’IEX existent: (1) échangeuse d'anions (phase stationnaire chargée positivement qui se lie à des protéines chargées négativement) et (2) échangeuse de cations (phase stationnaire chargée négativement qui se lie à des protéines chargées positivement). La chromatographie échangeuse d'ions est couramment utilisée comme une étape intermédiaire dans une méthode de purification des protéines, mais elle peut donner une résolution élevée pour certaines protéines lorsqu'elle est utilisée plus tôt ou plus tard au cours de la purification.
Toutes les protéines présentent une charge nette qui dépend de la composition en acides aminés de la protéine ainsi que des modifications attachées de manière covalente. La charge nette d'une protéine est influencée par le pH du solvant dans lequel elle est dissoute, comme le solvant échange des ions d'hydrogène avec les protéines. Le point isoélectrique (pI) d'une protéine est le pH auquel la protéine n'a pas de charge nette. A un pH supérieur au pI, une protéine aura une charge nette négative, tandis qu'un pH inférieur au pI mènera à une charge nette positive. Ainsi, le pH du solvant peut être ajusté afin de faciliter la liaison à l’IEX ou de promouvoir l'élution d'une protéine liée.

Théoriquement, toutes les protéines peuvent se lier à la fois aux matrices d'échange de cations et d'échange d'anions, si le pH du tampon est ajusté en conséquence. Toutefois, pour la purification de protéines, la stabilité de la protéine est le facteur le plus important dans le choix de conditions de purification et, par conséquent, la colonne la plus appropriée pour la liaison de la protéine. Par conséquent, il est nécessaire de déterminer si les conditions nécessaires pour la liaison à une des formes de chromatographie échangeuse d'ions affectent la stabilité et la fonction des protéines. Typiquement, les conditions de liaison à un type d'échangeur d'ions sont plus appropriées pour une protéine particulière que l'autre.
La connaissance du point isoélectrique d'une protéine peut aider à déterminer le type le plus approprié de chromatographie échangeuse d'ions. Les outils en ligne sont disponibles pour calculer le pI théorique d'une protéin EXPASY. Ces calculs sont basés entièrement sur la séquence d'acides aminés d'une protéine et ne prennent pas en compte la structure tridimensionnelle de la protéine. Dans son état naturel, des résidus d'une protéine sont plus exposés que d'autres et, par conséquent, le pI réel et la charge de surface nette sont parfois différents de ceux déterminés théoriquement [17]. La distribution des acides aminés les uns par rapport aux autres peut aussi affecter le pI d'une protéine [18], et cela n'est pas pris en considération avec les calculs théoriques de pI. Certaines techniques permettent la détermination expérimentale du pI d'une protéine à l'état natif, comme l’isoélectrofocalisation électrophorétiques [19], l’isoélectrofocalisation capillaire [20], et une approche à haut débit basée sur la luminescence plus récente [21].
En règle générale, la liaison d'une protéine à l’IEX doit être déterminée par essais et erreurs, en utilisant des solvants avec une gamme de valeurs de pH pour déterminer le pH optimal pour la rétention des protéines. Un pH de solvant qui est d'environ une unité de pH loin du pI est généralement suffisant pour la liaison de protéine [22] ; mais, dans certains cas, un pH plus éloigné du pI est nécessaire [23].
La phase stationnaire de IEX est composée d'une matrice d'agarose à base de polymère inerte ou avec un groupe chargé lié de façon covalente. Les particules de matrice sont disponibles en plusieurs tailles, et peuvent être non poreuses ou peuvent contenir des pores de taille variable. Le choix de la matrice dépend de la capacité de liaison désirée, la résolution et la vitesse d'écoulement. Les tailles de particules plus petites offrent une résolution plus élevée, mais nécessitent des débits plus faibles et prennent plus de temps. Les matrices poreuses offrent une capacité de liaison supérieure aux matrices non poreuses. Dans une matrice non-poreuse, les protéines ne peuvent pas entrer dans la résine et, par conséquent, ce type de matrice offre une meilleure récupération de l'échantillon, une meilleure résolution et des temps d'exécution plus courts que les matrices poreuses. Une étude a montré que, pour la plupart des protéines, la résolution et la récupération sont similaires quand elles sont séparées par une matrice poreuse ou non-poreuse, mais pour des protéines de grande taille, les matrices poreuses provoquent une perte de résolution basée sur les effets d'exclusion de taille [24].

Les quatre groupes fonctionnels chargés les plus couramment utilisés dans l’IEX sont indiqués ci-dessous. Ceux-ci sont classés comme échangeurs fort ou faible, avec des échangeurs forts étant ionisés dans une gamme de pH plus large que les échangeurs faibles.
Échange d'anions (phase stationnaire chargée positivement) :
- ammonium quaternaire (Q) - échangeur d'anions fort
- diéthylaminoéthyle (DEAE) - échangeur d'anions faible
L'échange de cations (phase stationnaire chargée négativement) :
- méthyl sulfonate de (S) - échangeur de cations fort
- carboxyméthyl (CM), - échangeur de cations faible
Les échangeurs forts doivent être utilisés lorsque le pH requis pour la liaison est très acide ou basique (en supposant que ce pH soit également approprié pour maintenir la stabilité des protéines), comme les groupes fonctionnels restent chargés sur une gamme de pH plus large [25]. Les échangeurs faibles fournissent moins de rétention pour certaines protéines, ce qui permet leur élution à une force ionique plus faible [26]. Parce que la force ionique élevée peut affecter la stabilité de certaines protéines [27], les échangeurs faibles pourraient être plus appropriés pour des protéines qui ne nécessitent pas un pH extrême pour la liaison.
Dans la chromatographie d'échange d'ions, la phase stationnaire est d’abord équilibrée avec un tampon de faible force ionique. L'échantillon de protéines est ensuite chargé sur la phase stationnaire dans le même tampon de faible force ionique utilisé pour l'équilibrage de la colonne. La protéine liée est lavée abondamment avant l'élution avec un tampon de force ionique plus élevée ou, dans certains cas, un tampon avec un pH modifié (figure 6). Les contre-ions dans le tampon d'élution interagissent avec la phase stationnaire chargée, en déplaçant la protéine liée. Certains sels sont plus efficaces pour une utilisation en chromatographie échangeuse d'ions que d'autres, en raison de leur aptitude à déplacer protéines liées et leurs effets sur la stabilité des protéines [28]. En général NaCl ou KCl sont utilisés pour l'élution, avec les ions Na+ ou K+ qui servent de contre-cations dans la chromatographie d'échange de cations et Cl - servant de contre-anion dans la chromatographie d'échange d'anions. Alternativement, le pH du tampon peut être modifié pour réduire la charge des protéines et perturber son interaction avec la phase stationnaire. Pour les protéines liées à une phase stationnaire d'échange de cations, l'augmentation du pH du tampon conduira à une charge positive moindre sur la protéine, et à l'élution de la colonne. Pour les protéines liées à une phase stationnaire d'échange d'anions, la diminution du pH du tampon conduira à une charge négative moindre sur la protéine, et à l'élution de la colonne.
Alternativement, le pH du tampon peut être ajusté de telle sorte que la protéine d'intérêt ne se lie pas à la phase stationnaire d'échange d'ions tandis que les protéines contaminantes le font. Dans ce cas, la protéine d'intérêt est recueillie dans la phase d’écoulement de la colonne, alors que certaines protéines contaminantes sont éliminées par leur liaison à la phase stationnaire.
Comme avec d'autres méthodes chromatographiques, la chromatographie par échange d'ions peut présenter des problèmes lors de la détermination des conditions optimales pour la liaison aux protéines, l'élution de la protéine, et une résolution adéquate.
| Problème | Cause | Solution |
|---|---|---|
| La protéine ne se lie pas | colonne pas équilibrée | Faire passer plus de tampon d’équilibration sur la colonne et recharger la protéine sur la colonne |
| La force ionique du tampon de liaison est trop élevée | Diminuer la force ionique du tampon | |
| pH n’est pas assez éloigné du pI | Ajuster le pH du tampon (diminuer pour l’échange de cation et augmenter pour l’échange d’anion) | |
| la protéine n’est pas éluée | La force ionique du tampon d’élution est trop faible | Augmenter la force ionique |
| la protéine est agrégée sur la colonne | Ajuster les conditions du tampon pour une meilleure stabilité de la protéine | |
| Résolution faible | le débit est soit trop rapide soit trop lent | Ajuster le débit |
| La colonne n’a pas été lavée suffisamment | Laver la colonne avec un tampon de force ionique plus élevée, nettoyer la phase stationnaire en suivant les recommandations du fournisseur | |
| La protéine est agrégée sur la colonne | Ajuster les conditions du tampon pour une meilleure stabilité de la protéine | |
| La protéine a perdu son activité durant la procédure | La protéine est dénaturée ou agrégée sur la colonne | Ajuster les conditions du tampon pour une meilleure stabilité de la protéine |
| un cofacteur nécessaire à l’activité a été éliminé pendant la purification | Ajouter un cofacteur |
La chromatographie d'interaction hydrophobe (HIC) sépare les protéines en fonction de leur caractère hydrophobe, et est souvent utilisé comme une étape intermédiaire dans un système de purification. Les protéines sont liées à une phase stationnaire dans un tampon de force ionique élevée et, par conséquent, HIC peut typiquement être réalisée immédiatement après la chromatographie par échange d'ions, sans échange de tampon ou de dilution nécessaire. HIC est aussi couramment réalisée après une précipitation au sulfate d'ammonium, une procédure qui peut être utilisée pour éliminer rapidement les protéines par précipitation de certaines, mais pas la totalité, des protéines avec du sel. HIC est parfois applicable dans les premières étapes d'un procédé de purification ou en tant qu'étape finale de l'élimination des traces d’impuretés de la protéine d'intérêt.
La présence d'ions de sel en solution peut conduire à la dénaturation partielle d'une protéine et à l'exposition des résidus hydrophobes normalement enfouis. Les protéines qui se lient à la phase stationnaire adoptent leur conformation native lorsqu’un tampon avec une force ionique plus faible est ajouté. Cela diminue l'exposition des résidus hydrophobes qui peuvent interagir avec la phase stationnaire, et facilite l'élution de la colonne. Pour les protéines qui peuvent se replier spontanément par une diminution de la force ionique, HIC peut être une méthode chromatographique précieuse pour la purification.
En règle générale, la force ionique du tampon de liaison doit être aussi faible que possible pour lier la protéine d'intérêt, tout en empêchant sa précipitation. Si la force ionique nécessaire pour la liaison aux protéines est élevée et provoque la précipitation de la protéine d'intérêt, une force ionique plus faible peut être utilisée. Dans ce cas, la procédure de chromatographie peut être utilisée pour séparer la totalité des protéines de liaison de la protéine d'intérêt qui, elle, n’est pas retenue sur la colonne et est éluée dans les premières étapes de la purification.
Avant de charger l'échantillon sur la colonne, la phase stationnaire doit être équilibrée avec le tampon de force ionique élevée (le même tampon dans lequel l'échantillon de protéine sera charge sur la colonne). L'échantillon est ensuite chargé sur la colonne, la colonne est lavée intensivement, et la protéine est éluée avec un tampon de force ionique faible (figures 7 et 8).
La phase stationnaire utilisée pour la chromatographie d'interaction hydrophobe est composée d'une matrice de base faite d’agarose réticulé ou d’un copolymère synthétique. Un ligand alkyle ou aryle est ensuite conjugué à la matrice de base, fournissant une spécificité pour des molécules hydrophobes.
Types de groupe fonctionnel:
- Alkyle - une chaîne hydrocarbonée de différentes longueurs; souvent un groupe butyle ou octyle est utilisé. La capacité de liaison de la phase stationnaire est augmentée avec l'augmentation de longueur de chaîne alkyle [29]. Les groupes fonctionnels lient des protéines entièrement sur l’hydrophobicité de la protéine
- Aryle - un groupe fonctionnel dérivé d'un cycle aromatique; souvent un groupe phényle. Les groupes aryle offrent spécificité croissante, les protéines peuvent également interagir avec le groupe fonctionnel grâce à des interactions d'empilement de base.
Le type et la concentration de sel utilisés pour la liaison et l'élution doivent être déterminés empiriquement pour chaque protéine. De plus, le repliement et la fonction de la protéine doit être assurée après son élution.
Comme d'autres formes de chromatographie sur colonne, une procédure HIC optimale peut nécessiter beaucoup de d’ajustement. L’ajustement des conditions de tampon et de la phase stationnaire sont nécessaires pour chaque protéine pour assurer une séparation optimale.
| Problème | Cause | Solution |
|---|---|---|
| La protéine ne se lie pas à la colonne | La colonne n’a pas été équilibrée | Faire passer plus de tampon d’équilibration sur la colonne et recharger la protéine |
| La force ionique du tampon de liaison est trop faible | Augmenter la force ionique du tampon | |
| La protéine s’agrège à la force ionique utilisée | Diminuer la force ionique du tampon ou essayer un sel diffèrent | |
| La protéine n’est pas éluée | La force ionique du tampon d’élution est trop élevée | Diminuer la force ionique |
| La protéine est agrégée sur la colonne | Ajuster les conditions du tampon pour une meilleure stabilité de la protéine | |
| La rétention est trop élevée | Essayer une phase stationnaire différente qui offre moins de rétention | |
| Résolution faible | Le débit est soit trop élevé ou trop faible | Ajuster le débit |
| La colonne n’a pas été lavée suffisamment | Laver avec un tampon de force ionique plus faible, nettoyer la phase stationnaire en suivant les recommandations du fournisseur | |
| La protéine est agrégée sur la colonne | Ajuster les conditions du tampon pour une meilleure stabilité des protéines | |
| faible rétention sur la colonne | Essayer une phase stationnaire différente qui offre une meilleure rétention | |
| La protéine perd son activité pendant la procédure | La protéine est dénaturée ou agrégée | Ajuster les conditions du tampon pour une meilleure stabilité des protéines |
| La protéine n’est pas revenue à sa conformation native | Essayer la liaison avec un sel différent ou à une force ionique plus faible, ajouter des additifs permettant une meilleure stabilité de la protéine | |
| Un cofacteur nécessaire à l’activité a été éliminé pendant la purification | Ajouter le cofacteur |
La chromatographie d'exclusion de taille sépare les protéines par leur rayon hydrodynamique, une propriété déterminée à la fois par la taille et la forme de la molécule. Contrairement aux autres méthodes chromatographiques décrites précédemment, les protéines ne se lient pas à une phase stationnaire en SEC. Au lieu de cela, les protéines sont séparées par la vitesse à laquelle elles naviguent à travers une phase stationnaire inerte (Figure 9).
La SEC est une méthode valable le plus souvent utilisée dans les étapes finales d'un système de purification en raison de sa capacité à différencier les différentes espèces d'une protéine. Les espèces oligomérique [30], les protéines dépliées [31], et tronquées [32] peuvent toutes être séparées de la protéine native, tout en échangeant simultanément des composants du tampon. Souvent, SEC est utilisée comme une méthode d'échange de tampon plus rapide et plus fiable que la dialyse, car elle est compatible avec plusieurs solvants et nécessite moins de tampon. Un seul solvant est utilisé tout au long de la procédure SEC, et les phases stationnaires SEC disponibles sur le marché sont compatibles avec les tampons les plus couramment utilisés.
Le type de la phase stationnaire utilisée et la longueur de la colonne jouent un rôle critique dans la résolution de protéines séparées par SEC. Plusieurs types de phases stationnaires sont disponibles dans le commerce, et la meilleure option dépend de la masse moléculaire des protéines qui nécessitent une séparation et les conditions dans lesquelles la séparation sera effectuée.
| Type de phase stationnaire | Matrice | caractéristiques |
|---|---|---|
| Sephadex | dextran et epichlorohydrine réticulé |
|
| Sephacryl | allyl dextran et N, N -méthylène bis acrylamide réticulé |
|
| Sepharose | agarose réticulé |
|
| Superose | agarose hautement réticulé |
|
| Superdex | agarose et dextran réticulé |
|
Superdex est souvent le meilleur choix pour les procédures SEC typiques, car il est compatible avec la plupart des solvants et offre la résolution la plus élevée de toutes les phases stationnaires disponibles.
Comme avec d'autres méthodes chromatographiques, il y a des avantages et des inconvénients à la SEC, et la décision d'utiliser cette méthode dépend à la fois de la protéine et de son application en aval.
Avantages de la SEC:
- Permet l’échange de tampon et le dessalage.
- Permet la séparation des espèces similaires (par exemple, les formes tronquées et oligomères) qui pourraient ne pas être séparées avec d'autres techniques de purification.
- Est compatible avec de nombreux solvants.
- Ne dépend pas de n'importe quelle propriété de protéine spécifique pour la rétention et l'élution.
Inconvénients de la SEC:
- La performance est très sensible à la préparation de la colonne.
- Peut présenter des interactions non spécifiques entre protéines et résine, ce qui diminue la résolution.
- Offre une faible résolution pour les mélanges complexes de protéines.
- L'échantillon doit être chargé dans un petit volume pour une résolution adéquate. Cela peut être problématique pour des protéines qui précipitent à des concentrations élevées.
L’optimisation des conditions de SEC pour atteindre la résolution maximale prend souvent beaucoup de temps, mais certains facteurs peuvent avoir un impact énorme sur la résolution.
Conditions qui augmentent la résolution des protéines en SEC :
- Réduire au minimum le volume de l'échantillon. Plus le volume est faible, moins diffuses seront les fractions éluées.
- Ajouter du sel dans le tampon. Une petite quantité de sel aidera à prévenir les interactions non spécifiques entre la protéine et la phase stationnaire. Cela permettra à toutes les protéines de passer au travers de la longueur de la colonne de manière consistante.
- Utilisation d'un débit modéré. Un débit trop rapide ne donne pas le temps pour les petites molécules de pénétrer dans les pores de la matrice, alors qu’un débit trop lent donne plus de temps pour la diffusion de l'échantillon.
- Veiller à ce que la viscosité de l‘échantillon et des solvants soient similaires. Ajuster les conditions de l'échantillon pour correspondre approximativement à celle du tampon.
- Réglez la longueur de la colonne. Une colonne qui est trop courte ne permet pas la séparation adéquate des protéines. Alors qu’une colonne qui est trop longue conduira à la diffusion de l'échantillon de protéine.
- Remplissage de la colonne. La préparation de la colonne peut avoir un effet énorme sur la résolution des protéines.
Si les particules ne sont pas bien dispersées ou si des bulles d'air sont emprisonnées dans la colonne alors le passage de l’échantillon à travers la phase stationnaire ne se fera pas correctement. De plus, si la colonne est autorisée à fonctionner à sec, elle doit être remplie à nouveau. Un mauvais garnissage de la colonne est souvent à blâmer pour une résolution faible inexplicable.
Parce que SEC ne sépare pas les protéines sur la base d’interactions avec un groupe fonctionnel, toutes les protéines sont éluées dans les mêmes conditions et, par conséquent, la résolution ne peut être obtenue que pour des protéines de rayon hydrodynamique très différent. Par conséquent, SEC n'est pas une méthode chromatographique appropriée pour les étapes initiales d'un système de purification quand il y a beaucoup de protéines contaminantes. Cependant, SEC offre une méthode rapide et fiable pour éliminer les sels ou de petites molécules à partir d'un échantillon entre le début ou les étapes intermédiaires de la purification. Dans les dernières étapes de purification, où seules des traces de contaminants existent, SEC est une méthode utile pour l'isolement des protéines et l'échange dans le tampon de stockage.
Les protéines qui se lient à une phase stationnaire sont éluées par des conditions de solvant qui perturbent les interactions. Ces conditions d'élution varient en fonction du type de la chromatographie et des propriétés de la protéine d'intérêt. Il existe trois méthodes générales d'élution des protéines: élution en une étape, élution par étapes, et un gradient d'élution linéaire. La meilleure méthode à choisir dépend du type de chromatographie utilise et de la résolution requise.
- Une étape - une seule étape d’élution déplace toutes les protéines liées. Ce mécanisme fonctionne le mieux pour les procédures chromatographiques basées sur une interaction très spécifique (par exemple, la chromatographie d'affinité). L’élution en une étape n'offre pas de résolution, mais est idéale pour se débarrasser des contaminants très rapidement. Elle nécessite la connaissance préalable des conditions de tampon nécessaires pour le déplacement de la protéine d'intérêt.
- Par étapes - plusieurs élutions sont effectuées séquentiellement, avec des conditions plus strictes à chaque étape. Dans une élution par étape, le nombre de fractions recueillies est fonction du nombre de conditions d'élution séquentielles. L’élution par étapes fournit une meilleure résolution que l’élution en une étape, mais une résolution plus faible que le gradient d'élution linéaire.
- Gradient linéaire - plusieurs fractions consécutives sont rassemblées tandis que les conditions d'élution sont ajustées de façon linéaire. Un gradient linéaire offre la plus haute résolution pour la chromatographie échangeuse d'ions et la chromatographie d'interaction hydrophobe. En règle générale, un grand nombre de fractions consécutives sont collectées.
La chromatographie d'exclusion de taille ne nécessite aucune de ces méthodes d'élution, comme aucune interaction ne se produit entre la protéine et la phase stationnaire. La protéine est chargée sur la colonne, et un grand nombre de fractions sont recueillies jusqu'à ce que toutes les protéines soient éluées, sans altérer les conditions de tampon durant toute la procédure.
Idéalement, le tampon d'élution d’une colonne est compatible avec la colonne suivante, ce qui élimine la nécessité d'un échange de tampon ou de dialyse entre les étapes de purification.
Après chaque séparation chromatographique, les fractions doivent être analysées pour déterminer les fractions contenant la protéine d'intérêt et la pureté relative de chacune de ces fractions. Cette analyse est nécessaire après chaque étape pour décider quelles fractions doivent être combinées pour une utilisation ultérieure. Pour déterminer la pureté, un test qui peut mesurer la quantité d'une protéine spécifique par rapport à la quantité de protéine totale est requis. Les tests suivants sont couramment utilisés pour l'analyse de pureté:
Électrophorèse sur gel de polyacrylamide (SDS-PAGE) - un gel dénaturant qui sépare les protéines en fonction de leur taille. Des colorants disponibles dans le commerce permettent une représentation visuelle de toutes les protéines dans l'échantillon, en offrant une évaluation qualitative de la pureté de la protéine (figure 10). L’électrophorèse SDS-PAGE n'est pas idéale pour l'analyse à haut débit des fractions et peut prendre plusieurs heures, mais est le plus souvent utilisée car facile, peu coûteuse et adaptée à n'importe quelle protéine.
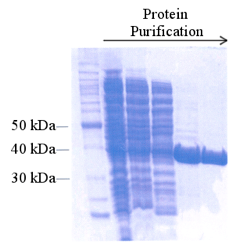 Figure 10. Électrophorèse SDS-PAGE d'échantillons collectés au cours d'un procédé de purification des protéines. Le gel est coloré pour la visualisation de toutes les protéines.de http://www.omicsonline.org/.
Figure 10. Électrophorèse SDS-PAGE d'échantillons collectés au cours d'un procédé de purification des protéines. Le gel est coloré pour la visualisation de toutes les protéines.de http://www.omicsonline.org/.- Spectroscopie - un procédé pour analyser les propriétés optiques des protéines. Cette technique d'analyse de la pureté de la protéine est seulement adaptée pour les protéines, telles que les cytochromes P450, qui ont une caractéristique spectroscopique unique. Les protéines de cette famille absorbent la lumière à une longueur d'onde là où d'autres protéines n’absorbent pas (autour de 420 nanomètres [33] ), de sorte que la comparaison de l'absorbance à 420 nm par rapport à 280 nanomètres (la longueur d'onde à laquelle toutes les protéines absorbent), peut fournir une mesure quantitative de pureté de la protéine. Cette méthode est rapide et à haut débit, mais uniquement pour certaines protéines.
- L'activité de protéine - un test enzymatique qui dépend de la protéine d'intérêt. Cette méthode d'évaluation de la pureté de la protéine est souvent associée à une autre forme de détermination de la concentration en protéines pour calculer l'activité par rapport à la concentration totale en protéines. Les dosages d'activité ne conviennent que pour des protéines ayant une activité qui peut facilement être mesurée dans un format à haut débit, telles que les protéases. Pour certaines protéines, des tests d'activité fournissent une méthode rapide et fiable pour la détection des protéines. La mesure de l'activité est souvent idéale pour les enzymes, parce que la protéine qui a perdu activité peut être exclue de toutes applications ultérieures.
Lorsque les protéines sont considérées comme suffisamment pures pour être utilisées dans des études expérimentales, elles doivent être entreposées de manière appropriée. La sélection d'un tampon de stockage final est tout aussi importante que le choix des tampons utilisés pendant la purification, et doit dépendre de la stabilité de la protéine et des conditions requises pour l'application en aval de la protéine purifiée. Souvent, la chromatographie par exclusion de taille est choisie en tant que dernière étape du procédé de purification, car le tampon de stockage peut être utilisé dans cette étape chromatographique efficace d'échange de tampon. Les fractions pures peuvent être mises en commun pour le stockage immédiat. Alternativement, les fractions finales regroupées peuvent être dialysées dans le tampon sélectionné pour le stockage.
Les conditions de stockage des protéines dépendent de la protéine d'intérêt et doivent être optimisées de sorte que la protéine maintienne sa stabilité structurale et fonctionnelle sur de longues périodes de stockage. Des additifs sont souvent inclus dans le tampon de stockage pour améliorer la durée de vie des protéines purifiées. Plusieurs essais sont souvent nécessaires pour déterminer les conditions optimales, comme toutes les protéines se comportent différemment.
Labome a revu 98 publications citant les méthodes de purification de protéines, avec un total de 242 exemples. La table 10 présente les principaux fournisseurs et les méthodes de purification. Les méthodes d’affinité et d’exclusion de taille sont les approches les plus couramment utilisées dans la littérature. GE Healthcare est le principal fournisseur pour toutes les méthodes, et Qiagen est le fournisseur prédominant pour la purification de protéines taguées avec le tag HIS, et Sigma Aldrich, pour la purification de protéines taguées avec le tag FLAG.
| type | Fournisseur | produit principal | num | Référence |
|---|---|---|---|---|
| Affinité | ||||
| HIS | Qiagen | Ni-NTA agarose | 26 | [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] [46] [47] [14] [48] [49] [50] [51] [52] [53] [7] [54] [55] [56] [57] |
| GE Healthcare | HisTrap FF/HP | 7 | [64] [65] [6] [66] [67] [68] [69] | |
| GE Healthcare | Ni Sepharose | 4 | [72] [73] [74] [45] | |
| Clontech | TALON Metal affinity | 7 | [76] [77] [78] [79] [80] [81] [82] [83] | |
| GST | GE Healthcare | glutathione Sepharose 4B | 14 | [84] [85] [86] [44] [79] [87] [78] [88] [41] [89] [53] [90] [91] [65] |
| Sigma-Aldrich | glutathione agarose | 2 | [94] [36] | |
| FLAG | Sigma-Aldrich | FLAG M2 affinity | 11 | [96] [89] [97] [67] [89] [98] [96] [97] [99] [67] [100] |
| D’autres systèmes de purification par chromatographie d’affinité ont également été cités comme Vector Laboratories agarose-bound Jacalin for 0-linked glycoproteins [81], New England Biolabs amylose resin for maltose-binding protein tag [36] [102] [76], Roche anti-Protein C affinity matrix [67], strepbiotin-based systems from Thermo Fisher Pierce [103] [76] [104], from EMD Millipore [106] [107], and from Sigma-Aldrich [103], Sigma-Aldrich Myc immunoaffinity resin [96], for metal ion-binding proteins, GE Healthcare Chelating Sepharose Fast Flow [108] aet GE Healthcare HiTrap IMAC HP column [109], heparin columns from GE Life Sciences [110] [111] [108], GE Healthcare Life Sciences HiTrap Protein A [14], GE Healthcare IgG Sepharose [112], et Sigma V5-agarose [90]. | ||||
| Exclusion de taille | ||||
| Superdex | GE-Healthcare | 31 | [114] [111] [109] [115] [69] [109] [45] [57] [73] [116] [46] [117] [14] [45] [118] [64] [49] [5] [38] [40] [73] [49] [54] [68] [7] [83] [119] [117] [74] [120] [72] [108], Superdex 200, 19 articles | |
| Superose | GE Healthcare | 4 | [46] [52] [54] [52] | |
| Sepharose | GE Healthcare | 2 | [102] [40] | |
| Echange d’ions | ||||
| anion | GE Healthcare | MonoQ column | 4 | [74] [87] [121] [55] |
| GE Healthcare | HiTrap Q | 4 | [122] [39] [48] [54] | |
| GE Healthcare | Source 15Q | 2 | [123] [36] | |
| Whatman | DE52 anion exchange resin | 1 | [110] | |
| cation | GE Healthcare | 1 | [46] | |
| Interactions hydrophobes | ||||
| GE healthcare | phenyl sepharose CL-4B | 1 | [41] | |
Parmi la littérature revue, les échantillons de protéines étaient préparés à l’aide de EMD Millipore BugBuster [73], [107], [50] ou AVESTIN EmulsiFlex C-3 cell disruptor [55] [124], et ont été concentrés à l’aide de EMD Millipore Amicon Ultra concentrators [120] [96] [110] [125] [81] ou deVivaspin concentrators [73] [122] [77] [120].
- Good N, Winget G, Winter W, Connolly T, Izawa S, Singh R. Hydrogen ion buffers for biological research. Biochemistry. 1966;5:467-77 PMID 5942950
- Grady J, Chasteen N, Harris D. Radicals from "Good's" buffers. Anal Biochem. 1988;173:111-5 PMID 2847586
- Desmarais W, Bienvenue D, Bzymek K, Holz R, Petsko G, Ringe D. The 1.20 A resolution crystal structure of the aminopeptidase from Aeromonas proteolytica complexed with tris: a tale of buffer inhibition. Structure. 2002;10:1063-72 PMID 12176384
- Ghalanbor Z, Ghaemi N, Marashi S, Amanlou M, Habibi-Rezaei M, Khajeh K, et al. Binding of Tris to Bacillus licheniformis alpha-amylase can affect its starch hydrolysis activity. Protein Pept Lett. 2008;15:212-4 PMID 18289113
- Pejchal R, Doores K, Walker L, Khayat R, Huang P, Wang S, et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science. 2011;334:1097-103 PMID 21998254 CrossRef
- Cruz-Migoni A, Hautbergue G, Artymiuk P, Baker P, Bokori-Brown M, Chang C, et al. A Burkholderia pseudomallei toxin inhibits helicase activity of translation factor eIF4A. Science. 2011;334:821-4 PMID 22076380 CrossRef
- Shenoy A, Wellington D, Kumar P, Kassa H, Booth C, Cresswell P, et al. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science. 2012;336:481-5 PMID 22461501 CrossRef
- Hernandez J, Stein A, Behrmann E, Riedel D, Cypionka A, Farsi Z, et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science. 2012;336:1581-4 PMID 22653732 CrossRef
- Mukherjee S, Behar M, Birnbaum H, Hoffmann A, Wright P, Ghosh G. Analysis of the RelA:CBP/p300 Interaction Reveals Its Involvement in NF-κB-Driven Transcription. PLoS Biol. 2013;11:e1001647 PMID 24019758 CrossRef
- Bohne A, Schwarz C, Schottkowski M, Lidschreiber M, Piotrowski M, Zerges W, et al. Reciprocal regulation of protein synthesis and carbon metabolism for thylakoid membrane biogenesis. PLoS Biol. 2013;11:e1001482 PMID 23424285 CrossRef
- Arakawa T, Philo J, Tsumoto K, Yumioka R, Ejima D. Elution of antibodies from a Protein-A column by aqueous arginine solutions. Protein Expr Purif. 2004;36:244-8 PMID 15249046
- Chong S, Mersha F, Comb D, Scott M, Landry D, Vence L, et al. Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene. 1997;192:271-81 PMID 9224900
- Jochum C, Beste M, Stone D, Graves S, Storb R. Development and in vitro characterization of canine CD40-Ig. Vet Immunol Immunopathol. 2008;123:260-5 PMID 18387675 CrossRef
- Corti D, Voss J, Gamblin S, Codoni G, Macagno A, Jarrossay D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850-6 PMID 21798894 CrossRef
- Shen Y, Tenney A, Busch S, Horn K, Cuascut F, Liu K, et al. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 2009;326:592-6 PMID 19833921 CrossRef
- Li F, Ravetch J. Inhibitory Fcγ receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333:1030-4 PMID 21852502 CrossRef
- Salaman M, Williamson A. Isoelectric focusing of proteins in the native and denatured states. Anomalous behaviour of plasma albumin. Biochem J. 1971;122:93-9 PMID 5124820
- Vesterberg O. Isoelectric fractionation, analysis, and characterization of ampholytes in natural pH gradients. V. Separation of myoglobins and studies on their electro-chemical differences. Acta Chem Scand. 1967;21:206-16 PMID 6031328
- Awdeh Z, Williamson A, Askonas B. Isoelectric focusing in polyacrylamide gel and its application to immunoglobulins. Nature. 1968;219:66-7 PMID 4173351
- Righetti P. Determination of the isoelectric point of proteins by capillary isoelectric focusing. J Chromatogr A. 2004;1037:491-9 PMID 15214685
- Pihlasalo S, Auranen L, Hanninen P, Harma H. Method for estimation of protein isoelectric point. Anal Chem. 2012;84:8253-8 PMID 22946671 CrossRef
- Ahamed T, Chilamkurthi S, Nfor B, Verhaert P, van Dedem G, van der Wielen L, et al. Selection of pH-related parameters in ion-exchange chromatography using pH-gradient operations. J Chromatogr A. 2008;1194:22-9 PMID 18154981
- Trodler P, Nieveler J, Rusnak M, Schmid R, Pleiss J. Rational design of a new one-step purification strategy for Candida antarctica lipase B by ion-exchange chromatography. J Chromatogr A. 2008;1179:161-7 PMID 18154980
- Duncan J, Chen A, Siebert C. Performance evaluation of non-porous versus porous ion-exchange packings in the separation of proteins by high-performance liquid chromatography. J Chromatogr. 1987;397:3-12 PMID 3654822
- Staby A, Jensen R, Bensch M, Hubbuch J, Dünweber D, Krarup J, et al. Comparison of chromatographic ion-exchange resins VI. Weak anion-exchange resins. J Chromatogr A. 2007;1164:82-94 PMID 17658538
- DePhillips P, Lenhoff A. Determinants of protein retention characteristics on cation-exchange adsorbents. J Chromatogr A. 2001;933:57-72 PMID 11758747
- von Hippel P, Wong K. On the conformational stability of globular proteins. The effects of various electrolytes and nonelectrolytes on the thermal ribonuclease transition. J Biol Chem. 1965;240:3909-23 PMID 5842063
- Tsumoto K, Ejima D, Senczuk A, Kita Y, Arakawa T. Effects of salts on protein-surface interactions: applications for column chromatography. J Pharm Sci. 2007;96:1677-90 PMID 17221853
- Er-El Z, Zaidenzaig Y, Shaltiel S. Hydrocarbon-coated sepharoses. Use in the purification of glycogen phosphorylase. Biochem Biophys Res Commun. 1972;49:383-90 PMID 4640365
- Woodbury R, Hardy S, Randall L. Complex behavior in solution of homodimeric SecA. Protein Sci. 2002;11:875-82 PMID 11910030
- Corbett R, Roche R. Use of high-speed size-exclusion chromatography for the study of protein folding and stability. Biochemistry. 1984;23:1888-94 PMID 6722129
- Kim T, Paik S, Yang C. Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry. 2002;41:13782-90 PMID 12427041
- Schenkman J, Jansson I. Spectral analyses of cytochromes P450. Methods Mol Biol. 2006;320:11-8 PMID 16719370
- Kralj J, Hochbaum D, Douglass A, Cohen A. Electrical spiking in Escherichia coli probed with a fluorescent voltage-indicating protein. Science. 2011;333:345-8 PMID 21764748 CrossRef
- Fritz J, Strehblow A, Taschner A, Schopoff S, Pasierbek P, Jantsch M. RNA-regulated interaction of transportin-1 and exportin-5 with the double-stranded RNA-binding domain regulates nucleocytoplasmic shuttling of ADAR1. Mol Cell Biol. 2009;29:1487-97 PMID 19124606 CrossRef
- Zalatan J, Coyle S, Rajan S, Sidhu S, Lim W. Conformational control of the Ste5 scaffold protein insulates against MAP kinase misactivation. Science. 2012;337:1218-22 PMID 22878499 CrossRef
- DiTacchio L, Le H, Vollmers C, Hatori M, Witcher M, Secombe J, et al. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science. 2011;333:1881-5 PMID 21960634 CrossRef
- Stefer S, Reitz S, Wang F, Wild K, Pang Y, Schwarz D, et al. Structural basis for tail-anchored membrane protein biogenesis by the Get3-receptor complex. Science. 2011;333:758-62 PMID 21719644 CrossRef
- Hirota T, Lee J, St John P, Sawa M, Iwaisako K, Noguchi T, et al. Identification of small molecule activators of cryptochrome. Science. 2012;337:1094-7 PMID 22798407 CrossRef
- Jinek M, Chylinski K, Fonfara I, HAUER M, Doudna J, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816-21 PMID 22745249 CrossRef
- Okada C, Yamashita E, Lee S, Shibata S, Katahira J, Nakagawa A, et al. A high-resolution structure of the pre-microRNA nuclear export machinery. Science. 2009;326:1275-9 PMID 19965479 CrossRef
- Heisel S, Ketter R, Keller A, Klein V, Pallasch C, Lenhof H, et al. Increased seroreactivity to glioma-expressed antigen 2 in brain tumor patients under radiation. PLoS ONE. 2008;3:e2164 PMID 18478111 CrossRef
- Tisserant A, König H. Signal-regulated Pre-mRNA occupancy by the general splicing factor U2AF. PLoS ONE. 2008;3:e1418 PMID 18183298 CrossRef
- McCloskey A, Taniguchi I, Shinmyozu K, Ohno M. hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science. 2012;335:1643-6 PMID 22461616 CrossRef
- Dueber E, Schoeffler A, Lingel A, Elliott J, Fedorova A, Giannetti A, et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science. 2011;334:376-80 PMID 22021857 CrossRef
- Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hintermair C, et al. CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science. 2012;336:1723-5 PMID 22745433 CrossRef
- Metcalf W, Griffin B, Cicchillo R, Gao J, Janga S, Cooke H, et al. Synthesis of methylphosphonic acid by marine microbes: a source for methane in the aerobic ocean. Science. 2012;337:1104-7 PMID 22936780 CrossRef
- Copic A, Latham C, Horlbeck M, D'Arcangelo J, Miller E. ER cargo properties specify a requirement for COPII coat rigidity mediated by Sec13p. Science. 2012;335:1359-62 PMID 22300850 CrossRef
- Diskin R, Scheid J, Marcovecchio P, West A, Klein F, Gao H, et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science. 2011;334:1289-93 PMID 22033520 CrossRef
- Zhang S, Bryant D. The tricarboxylic acid cycle in cyanobacteria. Science. 2011;334:1551-3 PMID 22174252 CrossRef
- Shi W, Zhang X, Jiang X, Yuan H, Lee J, Barry C, et al. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science. 2011;333:1630-2 PMID 21835980 CrossRef
- Breitsprecher D, Jaiswal R, Bombardier J, Gould C, Gelles J, Goode B. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science. 2012;336:1164-8 PMID 22654058 CrossRef
- Wang H, Westin L, Nong Y, Birnbaum S, Bendor J, Brismar H, et al. Norbin is an endogenous regulator of metabotropic glutamate receptor 5 signaling. Science. 2009;326:1554-7 PMID 20007903 CrossRef
- Armache K, Garlick J, Canzio D, Narlikar G, Kingston R. Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 Å resolution. Science. 2011;334:977-82 PMID 22096199 CrossRef
- Fleishman S, Whitehead T, Ekiert D, Dreyfus C, Corn J, Strauch E, et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816-21 PMID 21566186 CrossRef
- Wilusz J, Whipple J, Phizicky E, Sharp P. tRNAs marked with CCACCA are targeted for degradation. Science. 2011;334:817-21 PMID 22076379 CrossRef
- Klinge S, Voigts-Hoffmann F, Leibundgut M, Arpagaus S, Ban N. Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science. 2011;334:941-8 PMID 22052974 CrossRef
- Diao J, Burré J, Vivona S, Cipriano D, Sharma M, Kyoung M, et al. Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. elife. 2013;2:e00592 PMID 23638301 CrossRef
- Beyhan S, Gutiérrez M, Voorhies M, Sil A. A temperature-responsive network links cell shape and virulence traits in a primary fungal pathogen. PLoS Biol. 2013;11:e1001614 PMID 23935449 CrossRef
- Zimmermann I, Marabelli A, Bertozzi C, Sivilotti L, Dutzler R. Inhibition of the prokaryotic pentameric ligand-gated ion channel ELIC by divalent cations. PLoS Biol. 2012;10:e1001429 PMID 23185134 CrossRef
- Li Z, Park Y, Marcotte E. A Bacteriophage Tailspike Domain Promotes Self-Cleavage of a Human Membrane-Bound Transcription Factor, the Myelin Regulatory Factor MYRF. PLoS Biol. 2013;11:e1001624 PMID 23966832 CrossRef
- Yao P, Potdar A, Ray P, Eswarappa S, Flagg A, Willard B, et al. The HILDA Complex Coordinates a Conditional Switch in the 3'-Untranslated Region of the VEGFA mRNA. PLoS Biol. 2013;11:e1001635 PMID 23976881 CrossRef
- Walser R, Burke J, Gogvadze E, Bohnacker T, Zhang X, Hess D, et al. PKCβ Phosphorylates PI3Kγ to Activate It and Release It from GPCR Control. PLoS Biol. 2013;11:e1001587 PMID 23824069 CrossRef
- Lai Y, Cascio D, Yeates T. Structure of a 16-nm cage designed by using protein oligomers. Science. 2012;336:1129 PMID 22654051 CrossRef
- Hawley S, Fullerton M, Ross F, Schertzer J, Chevtzoff C, Walker K, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918-22 PMID 22517326 CrossRef
- Login F, Balmand S, Vallier A, Vincent-Monegat C, Vigneron A, Weiss-Gayet M, et al. Antimicrobial peptides keep insect endosymbionts under control. Science. 2011;334:362-5 PMID 22021855 CrossRef
- Puel A, Cypowyj S, Bustamante J, Wright J, Liu L, Lim H, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65-8 PMID 21350122 CrossRef
- King N, Sheffler W, Sawaya M, Vollmar B, Sumida J, Andre I, et al. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171-4 PMID 22654060 CrossRef
- Du J, Zhou Y, Su X, Yu J, Khan S, Jiang H, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806-9 PMID 22076378 CrossRef
- Lazarus J, Moughamian A, Tokito M, Holzbaur E. Dynactin subunit p150(Glued) is a neuron-specific anti-catastrophe factor. PLoS Biol. 2013;11:e1001611 PMID 23874158 CrossRef
- Iwig J, Vercoulen Y, Das R, Barros T, Limnander A, Che Y, et al. Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1. elife. 2013;2:e00813 PMID 23908768 CrossRef
- Huang N, Chelliah Y, Shan Y, Taylor C, Yoo S, PARTCH C, et al. Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science. 2012;337:189-94 PMID 22653727 CrossRef
- Azoitei M, Correia B, Ban Y, Carrico C, Kalyuzhniy O, Chen L, et al. Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science. 2011;334:373-6 PMID 22021856 CrossRef
- Barthelme D, Sauer R. Identification of the Cdc48•20S proteasome as an ancient AAA+ proteolytic machine. Science. 2012;337:843-6 PMID 22837385 CrossRef
- Schulz S, Iglesias-Cans M, Krah A, Yildiz O, Leone V, Matthies D, et al. A new type of na(+)-driven ATP synthase membrane rotor with a two-carboxylate ion-coupling motif. PLoS Biol. 2013;11:e1001596 PMID 23824040 CrossRef
- Meister S, Plouffe D, Kuhen K, Bonamy G, Wu T, Barnes S, et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science. 2011;334:1372-7 PMID 22096101 CrossRef
- Xu F, Wu H, KATRITCH V, Han G, Jacobson K, Gao Z, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322-7 PMID 21393508 CrossRef
- Oyama S, Yamakawa H, Sasagawa N, Hosoi Y, Futai E, Ishiura S. Dysbindin-1, a schizophrenia-related protein, functionally interacts with the DNA- dependent protein kinase complex in an isoform-dependent manner. PLoS ONE. 2009;4:e4199 PMID 19142223 CrossRef
- Org T, Chignola F, Hetényi C, Gaetani M, Rebane A, Liiv I, et al. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep. 2008;9:370-6 PMID 18292755 CrossRef
- Suzuki G, Shimazu N, Tanaka M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science. 2012;336:355-9 PMID 22517861 CrossRef
- Inamori K, Yoshida-Moriguchi T, Hara Y, Anderson M, Yu L, Campbell K. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science. 2012;335:93-6 PMID 22223806 CrossRef
- Paige J, Wu K, Jaffrey S. RNA mimics of green fluorescent protein. Science. 2011;333:642-6 PMID 21798953 CrossRef
- Brohawn S, del Mármol J, MacKinnon R. Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science. 2012;335:436-41 PMID 22282805 CrossRef
- Salahudeen A, Thompson J, Ruiz J, Ma H, Kinch L, Li Q, et al. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science. 2009;326:722-6 PMID 19762597 CrossRef
- Long D, Raschle M, Joukov V, Walter J. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333:84-7 PMID 21719678 CrossRef
- Sims R, Rojas L, Beck D, Bonasio R, Schüller R, Drury W, et al. The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science. 2011;332:99-103 PMID 21454787 CrossRef
- Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, et al. Function and molecular mechanism of acetylation in autophagy regulation. Science. 2012;336:474-7 PMID 22539722 CrossRef
- Bereczki O, Ujfaludi Z, Pardi N, Nagy Z, Tora L, Boros I, et al. TATA binding protein associated factor 3 (TAF3) interacts with p53 and inhibits its function. BMC Mol Biol. 2008;9:57 PMID 18549481 CrossRef
- Qian W, Miki D, Zhang H, Liu Y, Zhang X, Tang K, et al. A histone acetyltransferase regulates active DNA demethylation in Arabidopsis. Science. 2012;336:1445-8 PMID 22700931 CrossRef
- Fine B, Hodakoski C, Koujak S, Su T, Saal L, Maurer M, et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science. 2009;325:1261-5 PMID 19729658 CrossRef
- Pan F, Yu H, Dang E, Barbi J, Pan X, Grosso J, et al. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science. 2009;325:1142-6 PMID 19696312 CrossRef
- Xiong B, Bayat V, Jaiswal M, Zhang K, Sandoval H, Charng W, et al. Crag is a GEF for Rab11 required for rhodopsin trafficking and maintenance of adult photoreceptor cells. PLoS Biol. 2012;10:e1001438 PMID 23226104 CrossRef
- Bujalka H, Koenning M, Jackson S, Perreau V, Pope B, Hay C, et al. MYRF Is a Membrane-Associated Transcription Factor That Autoproteolytically Cleaves to Directly Activate Myelin Genes. PLoS Biol. 2013;11:e1001625 PMID 23966833 CrossRef
- Johnson K, Zhu S, Tremblay M, Payette J, Wang J, Bouchez L, et al. A stem cell-based approach to cartilage repair. Science. 2012;336:717-21 PMID 22491093 CrossRef
- Kudryashev M, Stenta M, Schmelz S, Amstutz M, Wiesand U, Castaño-Diez D, et al. In situ structural analysis of the Yersinia enterocolitica injectisome. elife. 2013;2:e00792 PMID 23908767 CrossRef
- Yi W, Clark P, Mason D, Keenan M, Hill C, Goddard W, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337:975-80 PMID 22923583 CrossRef
- Chakraborty A, Wang D, Ebright Y, Korlann Y, Kortkhonjia E, Kim T, et al. Opening and closing of the bacterial RNA polymerase clamp. Science. 2012;337:591-5 PMID 22859489 CrossRef
- He Y, Li B, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303-7 PMID 21817016 CrossRef
- Naidu S, Friedrich J, Russell J, Zomerdijk J. TAF1B is a TFIIB-like component of the basal transcription machinery for RNA polymerase I. Science. 2011;333:1640-2 PMID 21921199 CrossRef
- Lamia K, Sachdeva U, DiTacchio L, Williams E, Alvarez J, Egan D, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437-40 PMID 19833968 CrossRef
- Moin S, Urban S. Membrane immersion allows rhomboid proteases to achieve specificity by reading transmembrane segment dynamics. elife. 2012;1:e00173 PMID 23150798
- Wild P, Farhan H, McEwan D, Wagner S, Rogov V, Brady N, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228-33 PMID 21617041 CrossRef
- Seth D, Hausladen A, Wang Y, Stamler J. Endogenous protein S-Nitrosylation in E. coli: regulation by OxyR. Science. 2012;336:470-3 PMID 22539721 CrossRef
- Kaiser C, Goldman D, Chodera J, Tinoco I, Bustamante C. The ribosome modulates nascent protein folding. Science. 2011;334:1723-7 PMID 22194581 CrossRef
- Zhang M, Wu P, Kelly F, Nurse P, Hang H. Quantitative control of protein S-palmitoylation regulates meiotic entry in fission yeast. PLoS Biol. 2013;11:e1001597 PMID 23843742 CrossRef
- Previs M, Beck Previs S, Gulick J, Robbins J, Warshaw D. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337:1215-8 PMID 22923435 CrossRef
- Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443-6 PMID 21680843 CrossRef
- Coles C, Shen Y, Tenney A, Siebold C, Sutton G, Lu W, et al. Proteoglycan-specific molecular switch for RPTPσ clustering and neuronal extension. Science. 2011;332:484-8 PMID 21454754 CrossRef
- Wu X, Zhou T, Zhu J, Zhang B, Georgiev I, Wang C, et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333:1593-602 PMID 21835983 CrossRef
- Pinheiro V, Taylor A, Cozens C, Abramov M, Renders M, Zhang S, et al. Synthetic genetic polymers capable of heredity and evolution. Science. 2012;336:341-4 PMID 22517858 CrossRef
- Wu C, Li T, Farh L, Lin L, Lin T, Yu Y, et al. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science. 2011;333:459-62 PMID 21778401 CrossRef
- Carter A, Cho C, Jin L, Vale R. Crystal structure of the dynein motor domain. Science. 2011;331:1159-65 PMID 21330489 CrossRef
- Farcas A, Blackledge N, Sudbery I, Long H, McGouran J, Rose N, et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. elife. 2012;1:e00205 PMID 23256043 CrossRef
- Polikanov Y, Blaha G, Steitz T. How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science. 2012;336:915-8 PMID 22605777 CrossRef
- Soon F, Ng L, Zhou X, West G, Kovach A, Tan M, et al. Molecular mimicry regulates ABA signaling by SnRK2 kinases and PP2C phosphatases. Science. 2012;335:85-8 PMID 22116026 CrossRef
- Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, et al. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 2012;337:1340-3 PMID 22903523 CrossRef
- Janda C, Waghray D, Levin A, Thomas C, Garcia K. Structural basis of Wnt recognition by Frizzled. Science. 2012;337:59-64 PMID 22653731 CrossRef
- Liu X, Bushnell D, Silva D, Huang X, Kornberg R. Initiation complex structure and promoter proofreading. Science. 2011;333:633-7 PMID 21798951 CrossRef
- Liu T, Liu Z, Song C, Hu Y, Han Z, She J, et al. Chitin-induced dimerization activates a plant immune receptor. Science. 2012;336:1160-4 PMID 22654057 CrossRef
- Miller A, Long S. Crystal structure of the human two-pore domain potassium channel K2P1. Science. 2012;335:432-6 PMID 22282804 CrossRef
- Ataide S, Schmitz N, Shen K, Ke A, Shan S, Doudna J, et al. The crystal structure of the signal recognition particle in complex with its receptor. Science. 2011;331:881-6 PMID 21330537 CrossRef
- Spatzal T, Aksoyoglu M, Zhang L, Andrade S, Schleicher E, Weber S, et al. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science. 2011;334:940 PMID 22096190 CrossRef
- Oldham M, Chen J. Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science. 2011;332:1202-5 PMID 21566157 CrossRef
- Gardner B, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333:1891-4 PMID 21852455 CrossRef
- Ayaz P, Ye X, Huddleston P, Brautigam C, Rice L. A TOG:αβ-tubulin complex structure reveals conformation-based mechanisms for a microtubule polymerase. Science. 2012;337:857-60 PMID 22904013 CrossRef
- McNulty N, Wu M, Erickson A, Pan C, Erickson B, Martens E, et al. Effects of Diet on Resource Utilization by a Model Human Gut Microbiota Containing Bacteroides cellulosilyticus WH2, a Symbiont with an Extensive Glycobiome. PLoS Biol. 2013;11:e1001637 PMID 23976882 CrossRef
- réactif